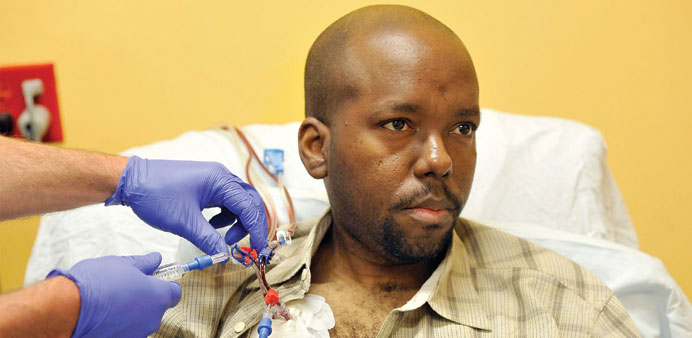
Dr Carlton Haywood, a faculty member at Johns Hopkins School of Public Health, has sickle cell and uses his own experience to teach others on ethical treatment of patients. He undergoes a monthly red blood cell exchange in Baltimore, Maryland.
By Jonathan Pitts
The tall, lanky patient enters the room bent over and shuffling like a man twice his age. He climbs on a gurney and lies back, head throbbing. Then Carlton Haywood pulls out a bottle of Tums, relief-in-waiting for the nausea he fears will come. “You never know how bad it’s going to get,” he says with a cordial smile.
Haywood, 37, belongs to the 0.003% of the US population that suffers from sickle cell disease, which predominantly affects blacks and which he has battled since birth. A healthcare team at the Johns Hopkins Outpatient Center is about to remove 75% of his blood and replace it with donated units, a procedure he undergoes monthly.
As rare as the blood disorder is, Haywood’s situation is even rarer. A bioethics and hematology professor at the Johns Hopkins University, he’s one of the few academicians who root a career studying the illness in personal experience.
Haywood the scholar knows Haywood the patient, a fact that imparts a special meaning and power to an often lonely mission. “There’s so much Carlton brings to our work that no one else can,” says Dr Sophie Lanzkron, the director of the Adult Sickle Cell Clinic at Hopkins. “He has significant illness, lives with this discomfort every day and manages it incredibly well. It’s one reason he knows as much about (sickle cell) as anybody I’ve met.”
On this June morning, Haywood reclines in fatigue as the bad blood flows from his body and the good enters through tubes in his chest. If he’s lucky, he’ll be well enough soon to return to a schedule of lecturing, writing and researching the state of sickle cell treatment in the US
On that last subject, he turns learned professor.
Most sufferers, Haywood says, have horror stories of locking horns with their healthcare providers. Many in the general population still believe the “black disease” they heard so much about 40 years ago has been cured. Worse, funding for research is 1/300ths of that available for comparable genetic illnesses.
“Sickle cell disease represents a uniquely bad confluence of problems,” he says in a voice far gentler than his disorder. Haywood pops a Tums.
“We have a long way to go, mm-hmm,” he says
Medical historians have traced sickle cell disease to four regions of Africa, where they say it developed thousands of years ago and was first described in medical literature in 1870. Today it affects about 100,000 people in the United States.
It strikes individuals of many ethnicities, including Caucasians, though more than 90% of sufferers are black. The medical system calls it an “orphan disease” — that is, like muscular dystrophy, cystic fibrosis and other disorders, it affects fewer than 200,000 people. But to those who have it, the numbers are more than big enough. And the problems start early.
Haywood was born in Atlanta, and for the first three years of his life, he screamed so often and so terribly that his parents, Carlton Sr, and Harriett Haywood, thought they must be doing something wrong. By 1979, when his sister Tammy was born, they had moved to Alabama, a state that mandated sickle cell testing for all newborns. She didn’t have the disease but carried some of its traits, so doctors suggested they test their son.
The results were “devastating,” says Harriett, now 61 and living in Columbia, South Carolina. “The doctors told us Carlton wouldn’t live through childhood. It was very, very difficult to deal with.”
The condition refers to misshapen red blood cells, according to the National Institutes of Health.
Normal red blood cells are doughnut-shaped and malleable, and contain hemoglobin, a protein that allows the cells to carry oxygen throughout the body. Hemoglobin-A — the normal kind — is ball-shaped, allowing the cells to move smoothly around even the sharpest turns in the body’s vascular system, reaching every organ.
Those born with sickle cell disease, though, inherit from both parents hemoglobin-S, which causes the cells to develop a crescent shape. The cells clump together and get stuck in curves and crannies, causing dangerous blockages that can last for hours.
As he explains the impact, a gentle smile crosses Haywood’s round face, his default expression whatever the subject. “During those crises, otherwise known as vaso-occlusive episodes, my organs are being starved of blood and oxygen,” he says.
The emergency is immediate — and has wide-ranging implications.
The pain is overwhelming, often so severe that only extremely powerful opioids can touch it. The affected organs can fail, sometimes fatally. And possibly most maddening, there exists no medical test to prove any attack is happening.
“Imagine you’re a 20-year-old African-American male in Baltimore City. You’re having intense pain. You go to the ER, where you’re appropriately pleading for narcotics, but you can’t prove anything is wrong,” says Lanzkron, who has treated Haywood as a patient and co-authored some of his research work. “You can imagine how that goes: often, not well.”
Haywood’s unsure why, but he always got fair treatment from physicians when growing up. It wasn’t until later that he realized how lucky that was. Sickle cell disease is so painful, complicated and poorly understood that for generations, medicine men in certain parts of Africa believed it was a ‘curse’.
Many Western patients use that word to describe it, Haywood says. He doesn’t blame them. He remembers childhood as a blur of pain crises, breathing tubes and oxygen masks. The weeks he spent in hospitals were lonely and frightening.
The episodes struck 10 or 12 times a year, usually starting in his chest or arms and spreading like a slow-burning fire to other parts of his body.
“Think of the worst pain you’ve ever had — a broken bone or a migraine — and imagine it as a steady rainfall,” he says. “Then it blossoms into a thunderstorm that lasts for hours.”
Nothing could kill the pain, but doctors prescribed morphine and other opioids to reduce it — just enough to bring relief without causing organ failure. The discomfort went beyond the physical.
Like many boys, Haywood loved sports, and he inherited a competitive nature from his father, the first African-American to win a full football scholarship to the University of South Carolina. But those who have sickle cell must monitor their activity. Overexertion, like dehydration, extreme weather, poor diet and stress, can spark attacks.
He landed in the emergency room so often that his mother drew a line. “I told him, ‘If you can’t strike a balance, I’m going to keep you in the house,’ “ she recalls. Between the ages of 11 and 14, he watched through a window as his friends played ball.
He threw himself into reading: science fiction, Stephen King and especially comic books, his preference during hospital stays. His parents discouraged him from delving into sickle cell, but he couldn’t help it. What he learned shocked him.
“Thinking about dying (as a child) is a nightmare,” Haywood says. He’d cry and scream and ask his parents, “Why?” They didn’t know, they said, but their answers gave him a bit of hope.
Carlton Sr, a manager for a credit company, taught him statistics. “Those are broad figures,” he’d say. “If 17 is the median (life span), some people live longer.”
“Everyone has an issue they’re dealing with,” said Harriett, a homemaker and devout Baptist. “This is yours. The question is, how will you deal with it?” Both told him to trust God.
They fetched his school assignments and made sure he kept up when he was out sick. He became a straight-A student. When he was 13, a doctor in yet another emergency room asked what he wanted to be when he grew up. Carlton wasn’t sure, but he did know that many comic-book superheroes were born of physics accidents.
“A physicist,” he said. “Well, you already know more about sickle cell than I do,” the doctor replied. “What about medicine?”
Years later, Haywood narrows his eyes at the memory. “Isn’t it interesting,” he says, “how a well-timed question can change the way you view what you might be able to do?”
Sickle cell overwhelmingly affects the urban poor, a demographic that suffers disproportionately from social instability, many studies show.
That means, among other things, that they often lack support when crises come.
As Haywood grew older, he realised he’d never had to drop out of school as so many with sickle cell do. He’d never had the disabling strokes that ruin school careers and wipe out futures by the thousands. His grades got him into the University of Virginia, where his plan was to pursue a career in medicine.
Harriett had always told him God gave him sickle cell for a reason. The idea used to make him angry. Now he wondered whether she had a point.
The attacks never went away — the day before his freshman year began, he suffered his worst crisis ever, causing him to miss a semester — but once he enrolled, even failure seemed to give him direction.
Early that first year, for instance, when he realised he disliked working in laboratories, an adviser suggested a subject he’d never heard of.
“Intro to Bioethics changed my life,” Haywood says. “I loved reading about ethical dilemmas in medicine. It presented a whole new way of asking questions.”
He earned a master’s degree in bioethics from Virginia and his doctorate in public health from Hopkins. Along the way, he saw as few others could that sickle cell, a disease at the molecular level, radiates into the world.
Take socioeconomics. Most patients, he learned, receive government medical assistance, which means their healthcare providers are undercompensated. This discourages others from going into the field, one reason so few primary-care physicians know the rudiments of sickle cell.
That problem reinforces another: lacking knowledge, healthcare providers are less likely to empathise with the patients’ unique needs, causing poorer treatment and more stress.
Worse, he found, funding for sickle cell research is abysmal. In 2004, Haywood came across a federally supported study that compared how well the US funds research into two comparable genetic disorders: sickle cell disease and cystic fibrosis. The results shocked him.
In 2003, it turned out, sickle cell disease attracted about $500,000 in private charitable funding. Cystic fibrosis, which affected fewer than a third as many people (nearly all of them Caucasian) attracted $150mn — 300 times as much.
That year, there were also 115 federally funded comprehensive care centers dedicated to cystic fibrosis. There were 10 for sickle cell disease according to the study, which later appeared in the journal Pediatrics.
Experts say the figures haven’t changed much. Sickle cell, it’s clear, is an orphan disease in more ways than one.
“The disparities are jaw-dropping,” says Lanzkron, who in 2008 founded the Sickle Cell Infusion Center at Hopkins, the only facility in Maryland that treats both children and adult patients. “The reasons, of course, are complicated. But when you show people these figures, it stuns them. We’re doing our best to spread the word.”
After two hours in a small room at HATS — the Hemaphaeresis and Transfusion Support Center at Hopkins Hospital — Haywood has had little nausea, a common side-effect of his treatment.
Dr Jay Brooks Jackson, a pathologist, and three nurses are draining most of Haywood’s blood and replacing it with 15 fresh units — about 1.18 gallons, or more than the average person has in his or her body.
In three weeks, he’ll have generated enough of his own blood to need a repeat.
Relieved that the end of the treatment is near, he’s glad to talk shop. He says he wears three hats in his sickle cell life, those of patient, professor and advocate. He trades them with ease.
Haywood the patient says medicine has made uncertain progress in his lifetime. When he was a child, a patient’s median life span was 14 years and few lived to age 18. By 2006, all 50 states had adopted universal newborn screening, which means every sufferer can be identified and given penicillin daily to head off infections. That has boosted the median life span to about 45 years, and 96% live at least 18 years.
But the Food and Drug Administration has approved just one drug, hydroxyurea, for use against sickle cell, and while it can reduce complications and pain in some patients, the treatment regimen is so complex that few who can use it do. Bone-marrow transplants have cured about 200 people, but they’re so invasive that few patients qualify.
Haywood the professor says patients and healthcare providers are still mutually wary. He has studied, among other things, what factors contribute to mutual trust in sickle cell-related doctor-patient relationships and how that level of trust compares to those in other disease populations. (Answer: not well.)
Can the gap be traced to race? After all, most patients are black, and most healthcare providers are not. Lanzkron calls the question “the elephant in the room.” Haywood is delicate on the subject. “It would make little sense to exclude (race) as a possibility,” he says, adding that any firm conclusions are a long way off.
Haywood the advocate heads a monthly research group at Hopkins that looks at disparities in sickle cell disease care, travels often to share his findings and speaks to each new class at the medical school. Psychology professor Shawn Bediako of University of Maryland, Baltimore County, a friend and colleague, said Haywood is skilled at explaining his work in a way that is accessible.
“No matter if you are a molecular geneticist, a person with a bachelor’s degree, or someone who dropped out of high school, you ‘get’ what he is doing, and you know why it is important,” Bediako said.
Haywood hopes to stumble on a donor with deep pockets or a celebrity who can take up a cause that, for whatever reason, has zero A-list spokesmen and little national visibility.
Haywood the man? He’s single. His mother keeps tabs on him via cell phone (his parents are separated), and his comic-book collection is bigger than ever. But the illness that shapes him still lurks like a shadow.
He knows some patients live into their 70s and beyond these days, and he realises he has lived longer than some expected, given his case’s severity.
But some nights he dreams of being in a strange, colourless void, a place of nothingness, and he wakes up, heart pounding. He believes in God, he says, though maybe not enough. Next morning, he gets up, pops his pain meds and heads out the door. There are others like him who need help. “I’m a lucky man,” he says. — The Baltimore Sun/MCT